Calculating the priming & therapeutic dose
For patients with a body surface area (BSA) of 2.2 m2 or less, calculate the dose using actual BSA:
- Patients with a BSA greater than 2.2 m2 should receive a dose based upon a BSA of 2.2 m2
- Dose adjustments do not need to be calculated for weight changes of less than or equal to 20%
To calculate the priming and therapeutic doses, multiply the patient’s BSA by the KYPROLIS® dose for the specific treatment regimen:
- Patient’s body surface area (BSA; m2) x dose (mg/m2)
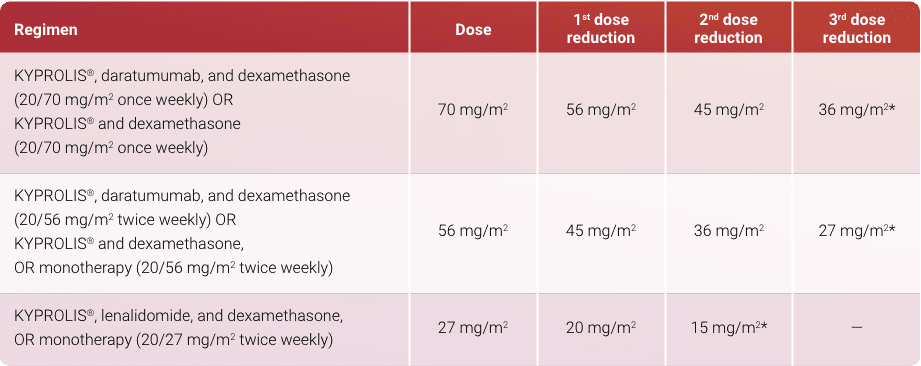
- Example: Calculate the correct Kd once-weekly dose for a patient with a BSA of 1.8 m2
Priming dose: 1.8 m2 x 20 mg/m2 = 36 mg
Therapeutic dose: 1.8 m2 x 70 mg/m2 = 126 mg
Using the patient’s total dose calculated, refer to the full Prescribing Information to determine the appropriate number of vials required for administration. KYPROLIS® is supplied in 60-mg, 30-mg, and 10-mg single-dose vials for reconstitution.
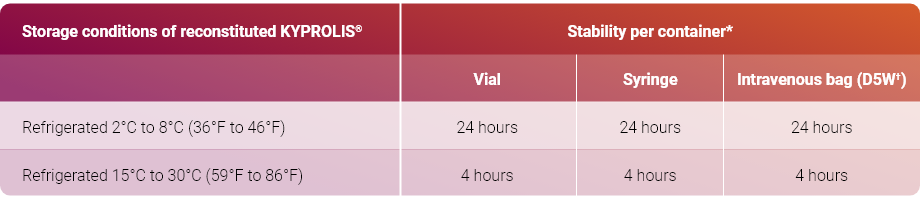
Read the complete preparation instructions prior to reconstitution. Parenteral drug products should be inspected visually for particulate matter and discoloration prior to administration, whenever solution and container permit.