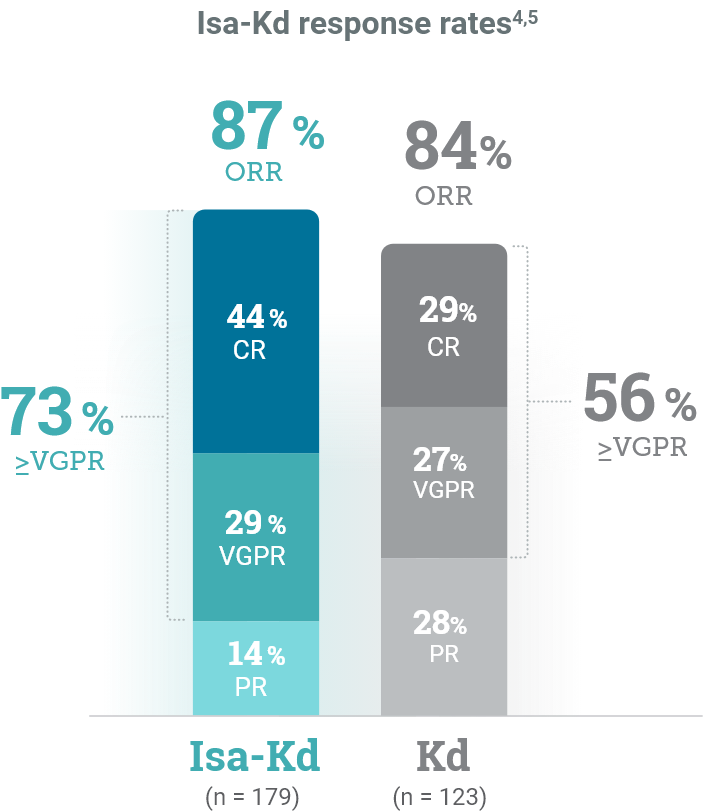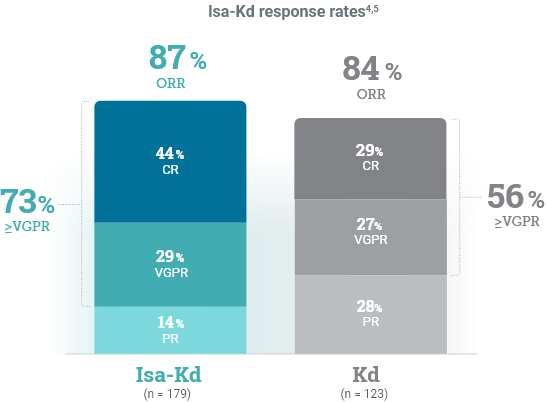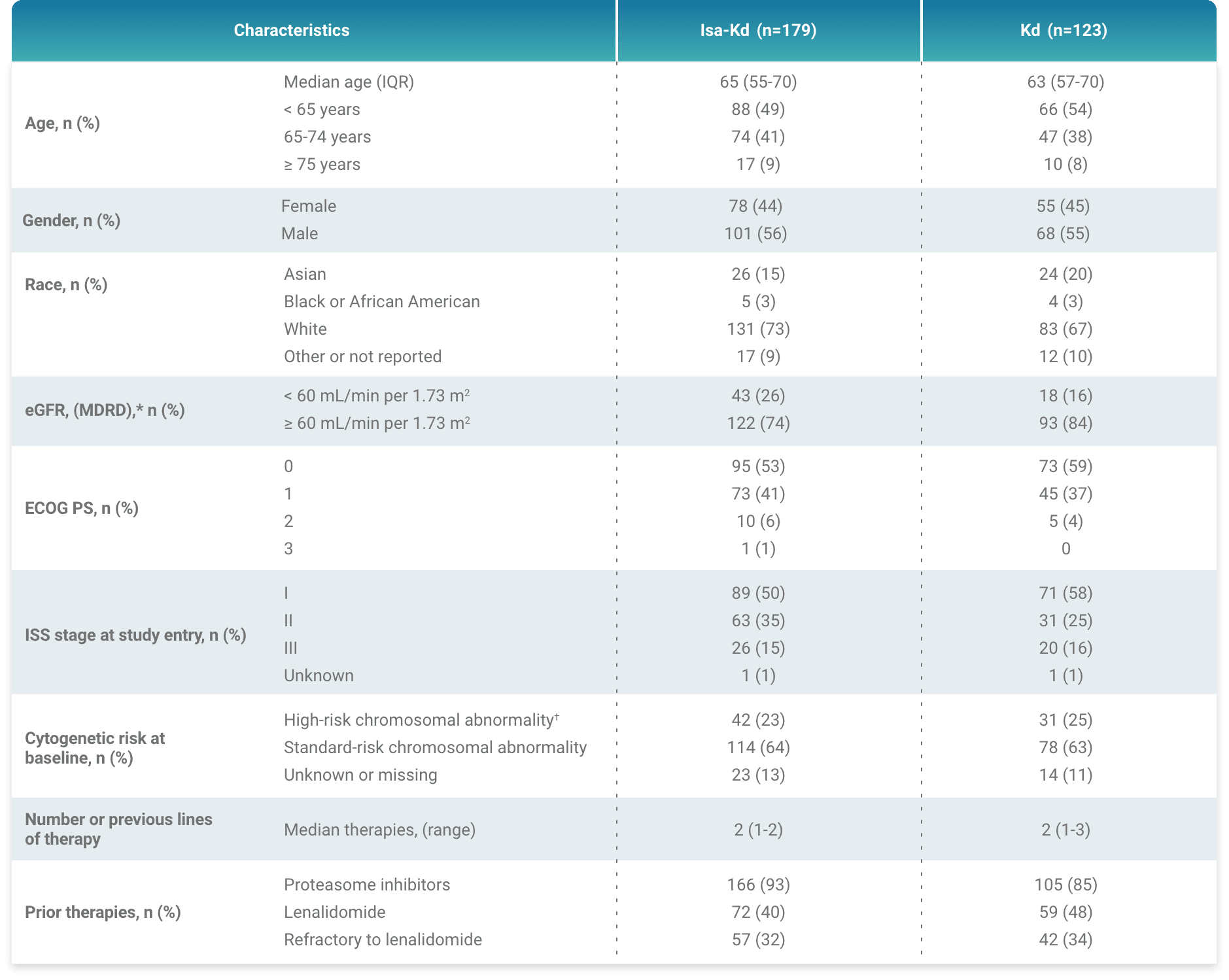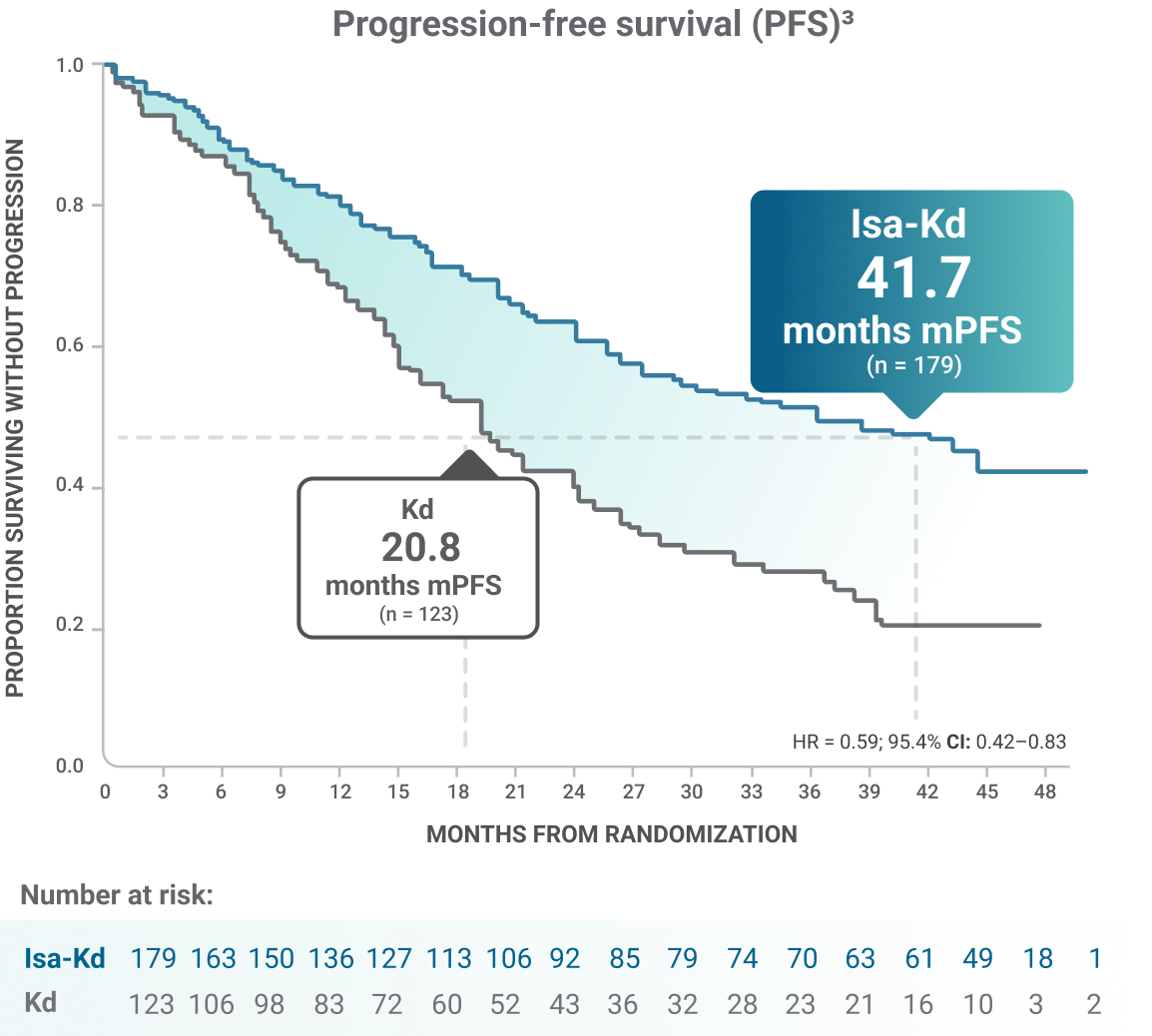

*As of the primary analysis, with a median follow-up of 20.7 months, the study met its primary endpoint of improved PFS. Median PFS was not reached with Isa-Kd vs 20.27 months with Kd (HR = 0.55; 95% CI: 0.37–0.82; P = 0.0032).
†Analysis censoring PFS events occurring >8 weeks from the last valid disease assessment.
CI, confidence interval; HR, hazard ratio; Isa-Kd, carfilzomib + isatuximab-irfc + dexamethasone; Kd, carfilzomib + dexamethasone; mPFS, median progression-free survival; PFS, progression-free survival; RRMM, relapsed or refractory multiple myeloma.