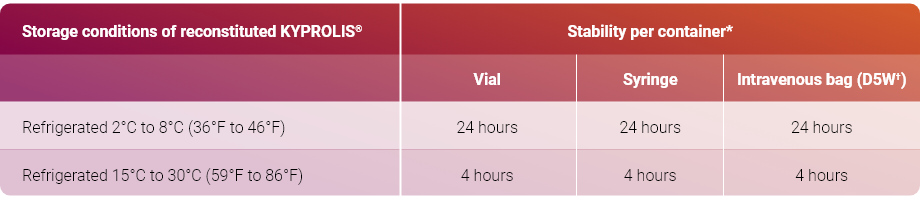
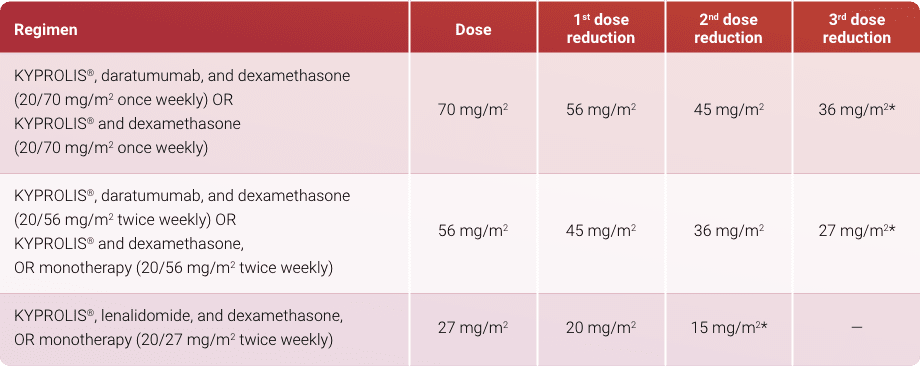
- KYPROLIS® (carfilzomib) is indicated in combination with dexamethasone, or with lenalidomide plus dexamethasone, or with daratumumab plus dexamethasone, ... Read More Close
INDICATIONS
Indications